Histoire La fibrose kystique : une histoire vieille de 34 000 ans
Les origines de la fibrose kystique sont lointaines. Plusieurs chercheurs ont tenté de découvrir le moment de l’apparition des principales mutations responsables de la maladie, entre autres grâce à l’analyse génétique de différentes populations concernées. En 2001, une étude a estimé l’âge de la mutation ∆F508, celle qui est la plus fréquente, entre 11 000 et 34 000 ans alors qu’une autre a estimé que l’apparition cette dernière pouvait même être antérieure à l’apparition des hommes modernes. Les autres mutations retrouvées dans la fibrose kystique sont légèrement plus récentes1.
La fibrose kystique, qui sera décrite scientifiquement pour la toute première fois comme une maladie à part entière en 1936, est observée depuis longtemps, du moins en ce qui a trait à ses effets et manifestations.
En 1595, une autopsie faite par le botaniste Pieter Pauw décrit une jeune fille rachitique de 11 ans, supposée ensorcelée par son entourage et présentant un pancréas élargi, dur et blanc. Puis, en 1606, le médecin espagnol Alonso Ruyzes de Fonteca rapporte que les doigts passés sur le front des enfants ensorcelés ont un goût salé5. Ce sont les premières traces officielles décrivant l’aspect de la maladie, bien qu’on ne la connaisse pas encore à l’époque et qu’on l’associe plutôt aux sorcières et au Diable. En effet, au Moyen Âge, on prédisait le sort funeste du nouveau-né dont la mère remarquait le « baiser salé », c’est-à-dire le goût salé laissé par un baiser sur le front de l’enfant 2, 3, 4. Un ancien proverbe en provenance de l’Europe du Nord était tristement répandu : « Malheur à l’enfant chez qui un baiser sur le front a un goût salé. Il est ensorcelé et doit mourir bientôt. »
Ce n’est qu’au début du 20e siècle qu’apparaissent les premières vraies observations scientifiques : en 1912, un chercheur décrit qu’il a observé des familles dont plusieurs enfants présentent une diarrhée graisseuse et meurent d’infection pulmonaire. Les
descriptions de l’époque se concentrent le plus souvent sur les problèmes digestifs et les troubles pancréatiques. Les médecins et chercheurs concluent en une forme de maladie cœliaque même si les problèmes bronchopulmonaires sont également notés et observés 6, 7, 8, 9.
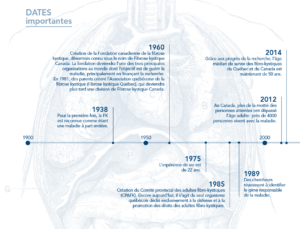
La fibrose kystique ne sera considérée comme une maladie à part entière qu’en 1938 par la pédiatre américaine Dorothy Andersen, médecin à l’Hôpital pour enfants de New York, qui publia alors un article intitulé « Fibrose kystique du pancréas et ses relations avec la maladie cœliaque »6. C’est en pratiquant des autopsies sur des nourrissons qu’elle découvrit les caractéristiques cliniques de la maladie, notamment l’obstruction intestinale néonatale, les complications respiratoires et digestives ainsi que les lésions histologiques spécifiques du pancréas. Elle relia cette maladie à un déficit en vitamine A10 et persista à soutenir cette théorie11, 12 pendant de nombreuses années. Bien qu’elle ait eu tort sur les causes de la maladie, nous savons aujourd’hui que le système digestif des personnes atteintes n’absorbe pas efficacement certains nutriments essentiels, comme les vitamines A, D, E et K. L’alimentation seule n’assurant pas un apport de vitamines suffisant pour combler les besoins de base, la prise de suppléments vitaminiques est souvent prescrite. Une des problématiques avec la maladie est la sécrétion insuffisante de certaines enzymes du pancréas, qui sont nécessaires à une bonne digestion. Cette situation nuit à l’absorption des matières grasses, des glucides et des protéines par l’organisme des personnes atteintes. C’est pourquoi, depuis déjà plusieurs décennies, les personnes atteintes de la FK ont presque tous recours à des suppléments d’enzymes lors de leurs repas13.
À l’été de 1948, une vague de chaleur frappe le nord des États-Unis. Celle-ci provoque alors l’hospitalisation de plusieurs enfants, qui souffrent de déshydratation et qui sont dans un état de léthargie avancée. Un médecin de l’hôpital général de New York, le Dr Paul di Sant’Agnesse, décide alors de comparer la composition de la sueur de ces enfants. En 1953, il découvre et explique les anomalies électrolytiques 14, 15 dans la sueur des malades, permettant d’envisager un diagnostic spécifique à la maladie, le test de la sueur. À cette époque, le diagnostic ne pouvait être fait que devant la combinaison de signes cliniques et de symptômes évocateurs d’insuffisance pancréatique et de malabsorption intestinale. Puis, on développe une technique au cours de laquelle les enfants étaient entièrement enveloppés dans des pansements, afin de stimuler la transpiration. Le test de la sueur devint le principal test pour établir clairement le diagnostic.
Dans les années 1950, le sort des enfants atteints est toujours considéré comme sans espoir, mais quelques centres spécialisés dans la prise en charge de la fibrose kystique voient le jour aux États-Unis et au Royaume-Uni. Le drainage postural est alors un des traitements traditionnels qui devient la norme. En 1955, on décrit de façon détaillée une méthode de prise en charge qui pose les fondements du traitement moderne : diagnostic précoce, traitement actif et précoce des infections pulmonaires, et surveillance et maintien de l’état nutritionnel 16. Lors de cette décennie, de nouveaux antibiotiques apparaissent. Alors que le Staphylococcus aureus est la bactérie principalement retrouvée, on note l’augmentation de la fréquence du Pseudomonas aeruginosa, attribuée aux traitements antibiotiques prolongés17. Par contre, le bénéfice de traitements antibiotiques agressifs devient progressivement évident18.
Au Canada, ce n’est qu’en 1969 qu’on voit apparaitre la première clinique spécifiquement dévouée à la fibrose kystique. Cette clinique voit le jour au Québec, à l’Hôpital Royal Edward de Montréal, qui deviendra par la suite l’Institut thoracique de Montréal. Il s’agit maintenant de la plus vieille clinique de FK au pays.
Au courant des années 1970, on décrit le rôle pathogène du Burkholderia cepacia, dont la dangerosité et la contagiosité entraînent des changements radicaux dans la pratique clinique et les habitudes sociales des patients 19. On se rend compte que la sévérité de l’infection par le P. aeruginosa et pronostic vital sont liés 20. Les progrès seront importants : la vision des médecins concernant la prise en charge de la maladie change progressivement.
En 1989, la découverte du gène défectueux de la fibrose kystique par Lap-Chee Tsui et John R. Riordan, de l’Hospital for Sick Children de Toronto, conduit à l’avancement actuel du savoir et du traitement de la maladie. La progression est majeure : l’anomalie génétique à l’origine de la maladie est enfin découverte. Cette découverte permettra par la suite
d’ajouter le génotypage au protocole diagnostique et d’envisager la thérapie génique. Il s’agit d’ailleurs du traitement le plus prometteur que l’on puisse envisager encore aujourd’hui.
Lors du congrès nord-américain sur la fibrose kystique tenu en 2014 à Atlanta, les chercheurs ont annoncé qu’il était désormais possible de corriger le gène défectueux. Bien que plusieurs recherches doivent toujours être effectuées, un traitement pouvant être administré à des fibro-kystiques présentant quelques mutations particulières est un avancement considérable. Il s’agit de l’ivacafto, qui permet le transport d’une quantité accrue de chlorure dans les cellules. En 2015, nous estimons qu’environ 130 patients Canadiens présentent les mutations pouvant être corrigées par ce médicament.
Bien qu’aucun remède pour guérir totalement la fibrose kystique n’existe encore, des méthodes efficaces de traitements sont disponibles. Actuellement, de plus en plus de jeunes atteignent l’âge adulte sans avoir subi trop de dommages aux poumons. La découverte du gène responsable de la fibrose kystique en 1989 promet aussi des méthodes de traitement encore plus perfectionnées. Grâce à la découverte de ce gène, les chercheurs peuvent retracer les liens entre la cause de cette maladie et l’ensemble de ses signes et symptômes.
Mucoviscidose ou fibrose kystique?
En 1938, suite à l’observation de ses jeunes patients malades, Dorothy Andersen utilise le terme Cystic fibrosis of the pancreas. Elle constate que ceuxci développent un kyste au niveau du pancréas. À l’époque et durant les années qui suivirent, les spécialistes ne feront pas un lien direct avec une maladie. Ce ne sera que bien des années plus tard qu’on établira le lien avec les autres problèmes, comme ceux affectant le système respiratoire. Ce sera en 1943 que le terme de mucoviscidosis, créé à partir des termes « mucus » et « visqueux », sera utilisé par le docteur Sydney Farber 21, un médecin américain, afin de corriger la dénomination employée par Dorothy Andersen, uniquement focalisée sur un organe, le pancréas. Farber étant alors persuadé que la maladie était plutôt due à une diffusion de mucus visqueux et épais dans plusieurs organes du corps : le terme de mucoviscidosis (mucoviscidose) se répandra rapidement dans le monde, particulièrement en Europe. Aux États-Unis et au Canada anglais, on utilise toujours le terme cystic fibrosis, alors qu’au Québec on préfère utiliser la première dénomination, soit fibrose kystique du pancréas.
Richard Leboeuf-mcGregor
Basé sur un article : Mucoviscidose. (2 novembre 2014). Wikipédia, l’encyclopédie libre.
Montréal (Québec) Canada
Références bibliographiques
1. American Lung Association. Racial/Ethnic Differences. Repéré à http://www.lungusa.org.
2. Emmanuelle Girodon-Boulandet,Catherine Costa, « Génétique de la mucoviscidose », Médecine thérapeutique / Pédiatrie, vol. 8, no 3, mai-juin 2005, p. 126-34
3. Scotet V, Duguépéroux I, Saliou P, Rault G, Roussey M, Audrézet MP, Férec C.« Evidence for decline in the incidence of cystic fibrosis: a 35-year observational study in Brittany, France »
4. Dr Thierry Bienvenu Fiche « Absence congénitale bilatérale des canaux déférents » Repéré à http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=FR&Expert=48
5. Alonso y de los Ruyzes de Fonteca, J. Diez Previlegios para Mgeres Preñadas. Henares, Spain: Alcalá de Henares, 1606, p. 212
6. Andersen DH, Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study. Am J Dis Child 1938; 56:344-399
7. Blackfan, K. D., and C. D. May. Inspissation of secretion, dilatations of the ducts and acini, atrophy and fibrosis of the pancreas in infants. J. Pediatr. 13: 627-634, 1938.
8. Harper, M. H.. Congenital steatorrhoea due to pancreatic defect. Arch. Dis. Child. 13: 45-56, 1938. Résumé [archive]
9. Fanconi G, Uehlinger E, Knauer C, « Das coeliakiesyndrom bei angeborener zysticher pankreasfibromatose und bronchiektasien » Wien Med Wschr 1936; 86:753-756.
10. Andersen DH. Cystic fibrosis of the pancreas, vitamin A deficiency and bronchiectasis. J Pediatr 1939; 15:763-771.
11. Andersen, Dorothy H. The Present Diagnosis and Therapy of Cystic Fibrosis of the Pancreas. Proc R Soc Med. 1949 Jan;42(1):25–32
12. Andersen DH. Therapy and prognosis of fibrocystic disease of the pancreas. Pediatrics 1949; 3: 406-417
13. Brunet. (2014). Les traitements de la fibrose kystique. Repéré à http://www.brunet.ca/fr/conseils/les-traitements-de-la-fibrose-kystique.html
14. Darling RC, diSant’Agnese PA, Perera GA, Andersen DH. Electrolyte abnormalities of the sweat in fibrocystic disease of the pancreas. A J Med Sci 1953; 225:67-70
15. Di Sant’ Agnese PA, Darling RC, Perera GA, et al. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas: clinical implications and relationship to the disease. Pediatrics 1953; 12:549–563
16. Jim Littlewood, « The history of the development of cystic fibrosis care. Perspective of a general paediatrician at a provincial teaching hospital. » [archive], surhttp://www.cysticfibrosismedicine.com [archive], The UK CFTrust, London, UK, 2002
17. Poncher, H Editor. Year Book of Pediatrics, 1951
18. Stoppelman MRH, Shwachman H. Effect of antibiotic therapy on mucoviscidosis: bacteriologic study. New Eng J Med 1954; 251:759-763.
19. Govan JR, Brown PH, Maddison J, Doherty CJ, Nelson JW, Dodd M, et al. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet 1993; 342:15-19
20. Hoiby N. Antibodies against Pseudomonas aeruginosa in serum from normal persons and patients colonised with mucoid or non-mucoid P. aeruginosa: results obtained by crossed immunoelectrophoresis. Acta Pathol Microbiol Immunol Scand 1977; 85:142-148
21. Farber S. Pancreatic insufficiency and the celiac syndrome. N Engl J Med 1943; 229:653-682Nike Air Max 98