History Cystic Fibrosis: a 34,000 year old story
An overview of the history of cystic fibrosis, from early ancient observations to modern medical advances.
The origins of cystic fibrosis are remote. Several researchers have tried to discover the time of the appearance of the main mutations responsible for the disease, among other things through the genetic analysis of different populations concerned. In 2001, one study estimated the age of the ∆F508 mutation, the most common one, between 11,000 and 34,000 years, while another estimated that the latter's appearance may even predate the appearance of modern humans. The other mutations found in cystic fibrosis are slightly more recent1.
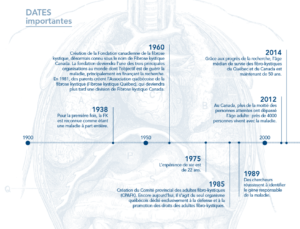
Cystic fibrosis, which was described scientifically for the very first time as a disease in its own right in 1936, has been observed for a long time, at least in terms of its effects and manifestations.
In 1595, an autopsy carried out by the botanist Pieter Pauw described a rickety 11-year-old girl, presumed to be bewitched by those around her and presenting an enlarged, hard and white pancreas. Then, in 1606, the Spanish physician Alonso Ruyzes de Fonteca reported that fingers passed over the forehead of bewitched children taste5. These are the first official records describing the aspect of the disease, although it was not yet known at the time and was instead associated with witches and the Devil. Indeed, in the Middle Ages, the fatal fate of the newborn was predicted whose mother noticed the “salty kiss”, that is to say the salty taste left by a kiss on the child's forehead 2, 3, 4. An ancient proverb from Northern Europe was sadly common: “Woe to the child for whom a kiss on the forehead has a salty taste. He is bewitched and must die soon.”
It was only at the beginning of the 20th century that the first real scientific observations appeared: in 1912, a researcher described that he observed families in which several children had fatty diarrhea and died from lung infections. Les
descriptions of the time most often focus on digestive problems and pancreatic disorders. Doctors and researchers conclude in a form of celiac disease even though bronchopulmonary problems are also noted and observed 6, 7, 8, 9.
Cystic fibrosis was not considered a disease in its own right until 1938 by the American pediatrician Dorothy Andersen, a doctor at the Children's Hospital in New York, who then published an article entitled “Cystic fibrosis of the pancreas and its relationship to celiac disease” 6. It was by performing autopsies on infants that she discovered the clinical characteristics of the disease, including neonatal intestinal obstruction, respiratory and digestive complications, and specific histological lesions of the pancreas. She linked this condition to vitamin A10 deficiency and persisted in supporting this theory11,12 for many years. Although she was wrong about the causes of the disease, we now know that the digestive system of affected people does not effectively absorb certain essential nutrients, such as vitamins A, D, E, and K. Because diet alone does not provide enough vitamins to meet basic needs, vitamin supplements are often prescribed. One of the problems with the disease is the insufficient secretion of certain pancreatic enzymes, which are necessary for good digestion. This situation interferes with the absorption of fats, carbohydrates and proteins by the body of affected people. This is why, for several decades now, people with CF have almost all been using enzyme supplements with their meals.13
In the summer of 1948, a heatwave hit the northern United States. This then causes the hospitalization of several children, who suffer from dehydration and who are in a state of advanced lethargy. A doctor at New York General Hospital, Dr. Paul di Sant'Agnesse, then decided to compare the composition of the sweat of these children. In 1953, he discovered and explained the electrolyte abnormalities 14, 15 in the sweat of patients, making it possible to consider a diagnosis specific to the disease, the sweat test. At that time, the diagnosis could only be made when there was a combination of clinical signs and symptoms suggestive of pancreatic insufficiency and intestinal malabsorption. Then, a technique was developed in which the children were completely wrapped in bandages to stimulate perspiration. The sweat test became the main test to establish a clear diagnosis.
In the 1950s, the fate of affected children was still considered hopeless, but a few centers specialized in the management of cystic fibrosis emerged in the United States and the United Kingdom. Postural drainage is then one of the traditional treatments that is becoming the norm. In 1955, a management method was described in detail that laid the foundations of modern treatment: early diagnosis, active and early treatment of lung infections, and monitoring and maintaining nutritional status 16. During this decade, new antibiotics appeared. While Staphylococcus aureus is the bacteria mainly found, there is an increase in the frequency of Pseudomonas aeruginosa, attributed to prolonged antibiotic treatments17. On the other hand, the benefit of aggressive antibiotic treatments is gradually becoming obvious18.
In Canada, it was not until 1969 that the first clinic specifically dedicated to cystic fibrosis appeared. This clinic was founded in Quebec, at the Royal Edward Hospital in Montreal, which later became the Montreal Chest Institute. It is now the oldest CF clinic in the country.
During the 1970s, the pathogenic role of Burkholderia cepacia was described, whose dangerous and contagious nature led to radical changes in the clinical practice and social habits of patients 19. It is realized that the severity of P. aeruginosa infection and the prognosis of life are linked 20. The progress will be significant: the vision of doctors concerning the management of the disease is gradually changing.
In 1989, the discovery of the defective cystic fibrosis gene by Lap-Chee Tsui and John R. Riordan of the Hospital for Sick Children in Toronto led to the current advancement of knowledge and treatment of the disease. The progression is major: the genetic anomaly at the origin of the disease is finally discovered. This discovery will allow later
to add genotyping to the diagnostic protocol and to consider gene therapy. In fact, it is the most promising treatment that can still be considered today.
At the North American Cystic Fibrosis Congress held in 2014 in Atlanta, researchers announced that it was now possible to correct the faulty gene. Although much research still needs to be done, a treatment that can be given to cystic fibrosis with a few specific mutations is a considerable advance. It is ivacafto, which allows the transport of an increased quantity of chloride into cells. In 2015, we estimate that approximately 130 Canadian patients have mutations that can be corrected by this medication.
Although no cure for a complete cure for cystic fibrosis still exists, effective treatment methods are available. Currently, more and more young people are reaching adulthood without having suffered too much lung damage. The discovery of the gene responsible for cystic fibrosis in 1989 also promises even more advanced treatment methods. Thanks to the discovery of this gene, researchers can trace the links between the cause of this disease and all of its signs and symptoms.
Cystic fibrosis or cystic fibrosis?.
In 1938, following the observation of her young sick patients, Dorothy Andersen used the term Cystic Fibrosis of the Pancreas. She notes that they develop a cyst in the pancreas. At the time and in the years that followed, specialists would not make a direct link to a disease. It would be many years later that the link with other problems, such as those affecting the respiratory system, would be established. It was in 1943 that the term cystic fibrosis, created from the terms “mucus” and “viscous”, was used by Dr. Sydney Farber 21, an American doctor, in order to correct the name used by Dorothy Andersen, which was only focused on one organ, the pancreas. Farber was then convinced that the disease was rather due to a diffusion of viscous and thick mucus in several organs of the body: the term cystic fibrosis (cystic fibrosis) spread rapidly around the world, particularly in Europe. In the United States and English Canada, the term cystic fibrosis is still used, while in Quebec, the first name is preferred, which is pancreatic cystic fibrosis.
Richard LeBoeuf-McGregor
Based on an article: Cystic Fibrosis. (November 2, 2014). Wikipedia, the free encyclopedia.
Montreal (Quebec) Canada
Bibliographical references
1. American Lung Association. Racial/Ethnic Differences. Retrieved from http://www.lungusa.org.
2. Emmanuelle Girodon-Boulandet, Catherine Costa, “Genetics of cystic fibrosis”, Therapeutic Medicine/Pediatrics, vol. 8, no 3, May-June 2005, p. 126-34
3. Scotet V, Duguépéroux I, Saliou P, Saliou P, Rault G, Rault G, Roussey M, Audrézet MP, Férec C. “Evidence for decline in the incidence of cystic fibrosis: a 35-year observational study in Brittany, France”
4. Dr. Thierry Bienvenu Sheet “Bilateral congenital absence of vas deferens” Retrieved from http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=FR&Expert=48
5. Alonso y de los Ruyzes de Fonteca, J. Diez Previlegios para Mgeres Preñadas. Henares, Spain: Alcalá de Henares, 1606, p. 212
6. Andersen DH, Cystic fibrosis of the pancreas and its relationship to celiac disease: a clinical and pathological study. Am J Dis Child 1938; 56:344-399
7. Blackfan, K.D., and C.D. May. Inspiration of secretion, dilatations of the ducts and acini, atrophy and fibrosis of the pancreas in infants. J. Pediatr.13:627-634, 1938.
8. Harper, Mr. H. Congenital steatorrhoea due to pancreatic defect. Arch. Say. Child. 13:45-56, 1938. Summary [archive]
9. Fanconi G, Uehlinger E, Knauer C, “The coeliac syndrome at the management of systemic pancreatifibromatosis and bronchiectasis” Wien Med Wschr 1936; 86:753-756.
10. Andersen D. H. Cystic fibrosis of the pancreas, vitamin A deficiency and bronchiectasis. J Pediatrr 1939; 15:763-771.
11. Andersen, Dorothy H. The Present Diagnosis and Therapy of Cystic Fibrosis of the Pancreas. Proc R Soc Med. 1949 Jan; 42 (1) :25—32
12. Andersen D. H. Therapy and prognosis of fibrocystic disease of the pancreas. Pediatrics 1949; 3:406-417
13. Brunet. (2014). Treatments for cystic fibrosis. Retrieved from http://www.brunet.ca/fr/conseils/les-traitements-de-la-fibrose-kystique.html
14. Darling RC, saying Agnese PA, Perera GA, Andersen DH. Electrolyte abnormalities of the sweat in fibrocystic disease of the pancreas. A J Med Sci 1953; 225:67-70
15. Di Sant' Agnese PA, Darling RC, Perera GA, et al. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas: clinical implications and relationship to the disease. Pediatrics 1953; 12:549 —563
16. Jim Littlewood, “The history of the development of cystic fibrosis care. Perspective of a general paediatrician at a provincial teaching hospital.” [archive], at http://www.cysticfibrosismedicine.com [archive], The UK CFTrust, London, UK, UK, 2002
17. Poncher, H Editor. Year Book of Pediatrics, 1951
18. Stoppelman MRH, Shwachman H. Effect of antibiotic therapy on cystic fibrosis: bacteriologic study. New Eng J Med 1954; 251:759-763.
19. Govan JR, Brown PH, Maddison J, Maddison J, Doherty CJ, Doherty CJ, Nelson JW, Dodd M, et al. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet 1993; 342:15-19
20. Hoiby N. Antibodies against Pseudomonas aeruginosa in serum from normal persons and patients colonised with mucoid or non-mucoid P. aeruginosa: results obtained by crossed immunoelectrophoresis. Acta Pathol Microbiol Immunol Scand 1977; 85:142-148
21. Farber S. Pancreatic insufficiency and the celiac syndrome. N Engl J Med 1943; 229:653-682
Research
Antibiotic resistance and Pseudomonas virulence: two sides of the same coin
In people with cystic fibrosis, some resistance to antibiotics caused by Pseudomonas aeruginosa may paradoxically increase bacterial virulence, complicating lung infections and opening the way to new therapeutic approaches.
Thanks to Our Partners





